独立行政法人 産業技術総合研究所【理事長 野間口 有】(以下「産総研」という)ナノシステム研究部門【研究部門長 八瀬 清志】ダイナミックプロセスシミュレーショングループ 宮本 良之 研究グループ長は、シミュレーションに基づいて、パルス幅2フェムト秒(fs)程度のレーザー照射により、酸化グラフェンを還元してグラフェンを製造する方法を提案した。
時間依存第一原理計算によりフェムト秒レーザーを照射した後の酸化グラフェンの構造変化をシミュレーションすることにより、効率良く酸化グラフェンを還元してグラフェンを作製するのに適したレーザーの波形を見出した。この還元方法は、化学物質を用いたり、高温で処理したりしない方法で、パルス幅の広い(~200 fs)フェムト秒レーザーによる還元よりも、還元反応に伴う発熱を抑制できるため、グラフェンに欠陥が発生するリスクが少ない。したがって、この還元方法を応用すれば酸化グラフェンの印刷塗布によるグラフェン電極製造技術への貢献が期待される。
なお、本成果の詳細は2012年1月17日(日本時間)に米国物理学会発行のPhysical Review Bにオンライン掲載される。また2012年2月15~17日に東京ビッグサイト(東京都江東区)で開催されるnano tech 2012第11回 国際ナノテクノロジー総合展・技術会議にて発表する。
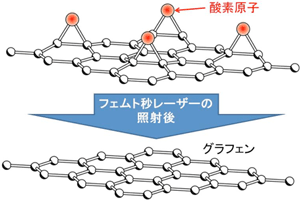 |
| レーザー照射により酸化グラフェンから酸素が除かれるシミュレーションの結果 |
近年、グラフェンを応用した電子技術が注目されている。透明性と電気伝導度がともに高いという性質を活かして、太陽電池用の電極やタブレットPCなどのタッチパネルへの透明電極としての応用が期待されている。しかし、グラフェンをこれらの用途に利用するために大量合成し、必要なパターンで印刷塗布する技術にはコストや結晶性劣化などの問題があり、開発の妨げになっていた。
近年、グラファイトを酸化して酸化グラフェンに変えた後、溶液中で剥離し、それを回路パターンに印刷塗布した後に還元するというグラフェン電極製造方法が注目されている。しかし還元には、ヒドラジンなどの毒性の強い化学物質を用いるか、1000 ℃の高温で処理する必要があり実用化の障害となっていた。
産総研は実用化に適したグラフェン応用技術の開発を目指しており、電気伝導度の計算によるエレクトロニクス材料としての検証、電界印加によるバンドギャップのコントロールによるトランジスタの設計、グラフェンを高性能で動作させるための基板の選択などの理論的研究も進めてきた。グラフェン研究、特に製造方法の研究は世界的に競争が激しい。そのなかで、酸化グラファイトを溶液中で剥離して酸化グラフェンを得、それを塗布後、還元することでグラフェンを製造する技術に着目し、酸化グラフェンの還元をより効率的に行う方法を探ることとした。
今回の研究は計算プログラムをともに開発してきた中華人民共和国 四川大学のHong Zhang教授との共同研究であり、文部科学省「HPCI戦略プログラム」および「計算物質科学イニシアティブ」の助成を受けたものである。計算には国立大学法人 筑波大学のT2Kスーパーコンピューターを利用し、計算の実行および解析は産総研が担当した。
今回の研究では、産総研の第一原理計算技術とその計算プログラムを活用して、レーザー光による酸化グラフェンの電子励起とそれに引き続いて起こる酸化グラフェン内の原子の運動のシミュレーションを行った。これは、時間依存密度汎関数理論に基づく第一原理計算によって、レーザー照射直後からの電子の波動関数の時間変化と原子核の分子動力学の計算を同時に実行することで可能となった。パルス波形をさまざまに変えてシミュレーションすることによって、酸化グラフェンにダメージを与えないで還元する方法に適したフェムト秒レーザーの波形を見出した。
さまざまな波形のうち、図1のような電場変化を示すパルス波形のレーザーが最も効率がよいことが判明した。このレーザー波形はパルス幅2 fsと、これまでに酸化グラフェンの還元に利用されているフェムト秒レーザー(パルス幅は約200 fs)よりも著しく狭いパルス幅である。
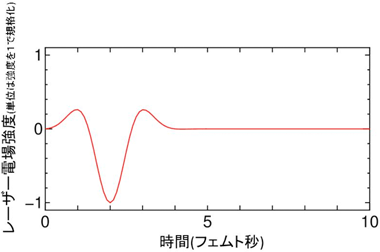 |
図1 第一原理計算で見出された、酸化グラフェンの還元に最も適した極短パルスレーザー波形
この波形は、レーザー光による電場の向きの時間変化の平均が非対称的であり、電場の向きが酸素原子の吸着した面より下向きになる時間が長くなっている。 |
今回のシミュレーションの条件では、レーザーから供給されるエネルギー密度がある閾値を超えると、図2に示すように、酸化グラフェンから酸素原子が脱離すること、すなわち還元されることが分かった。酸素原子の脱離運動が顕著になる時間スケールでは、レーザーパルスはもはや減衰しきっている。これは、電子励起が高速で起きるのに対し、原子運動が始まる時間は遅いためで、電子と原子の質量差によるものである。また、酸素原子との結合により歪んでいたグラフェンの炭素原子の配列が、酸素原子の脱離後にはグラフェン本来の平坦な構造に戻ることも計算により分かり、この方法による還元ではグラフェンの構造が破壊されないことが示された。還元に必要なレーザーのエネルギー密度の計算値は数 mJ/cm2のオーダーであることが本計算より分かったが、より精密な絶対値はシミュレーションのために想定した周期境界条件に依存するため、酸化グラフェンの還元を行うためのレーザーエネルギーの閾値をシミュレーションで精密に決定するには至っていない。
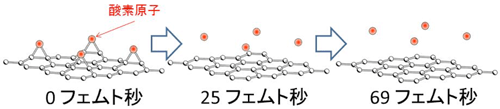 |
| 図2 グラフェンに結合した酸素原子(エポキシ構造を構成)が極短パルスレーザー照射後に脱離するシミュレーション結果 |
酸化グラフェンのもう1つの形態である水酸基(OH基)をもつ構造についてもシミュレーションを実行した。この場合も、同じ波形のパルスレーザー照射によりOH基が脱離し、酸化グラフェンが還元されることが判明した。図3はその様子をシミュレーションした結果である。
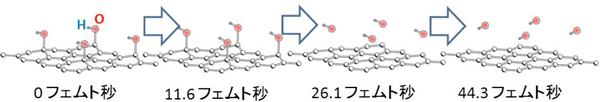 |
| 図3 酸化グラフェンからOH基がパルスレーザー照射後に脱離するシミュレーション結果 |
OH基の場合には、パルスレーザー照射後に、まず質量の軽い水素が高速運動を開始するが、水素は脱離せずそのままで、酸素が脱離を始める。このまま水素と酸素の結合は切れることはなく、水素は脱離の速度をいったん緩め、酸素に追いつかれる。このようにしてOH基は分子軸の方向を揺らしながらグラフェンから離れていくことが分かった。
電子のダイナミクスを計算する際に便宜上導入した周期境界条件の及ぼす影響のさらなる検証、酸化グラフェンのレーザー照射前の温度条件を統計的に取り込むなどの計算上の技術的問題から発生する計算精度の問題を解決することで、レーザー波形だけでなく、還元を行うために必要最小限度のレーザー強度の予測精度向上を目指す。グラフェンの生成時に酸化以外の原因による不純物除去の方法へと研究を拡大することで、グラフェン生成、精製のプロセスの精密設計が可能となり、実用化につながると期待している。